Getting Started with Examples
Getting_Started_with_Examples.RmdIt is highly recommended to begin by running the following example hand-in-hand with this guide
Download example log files
Please find the example log files on the package’s Github
Once downloaded and unzipped, you are ready to go!
For user convenience, we demonstrate usage with a small number of molecules, such that downloading the log files directly to a local machine will stay within memory-usage reason. As stated on the home page, it is generally not the case.
Run extRactoR
# Run extRactoR (no need to change working directory - extRactoR does it for you)
extRactoR()Choosing log files location
Choose the location (the directory and not a specific file) of the log files
Naming the directory
Name your directroy - this directory will contain the resulting .feather files.
Allowing moleculaR workflow
Unless it is a real deal breaker, allow moleculaR do its thing - will make your life easier (and mine)
In case you decide not to - answer “no” and choose a location that will make sense
This is the slowest part of the process, and it a takes a few seconds per molecule - be patient, if the program doesn’t collapse with an error message, it means its running!
Once done, you will receive a message on your console:
Done! Current working directory is set to path/to/example_log_files/Name_Chosen_By_User
Run unwRapper
# Run unwRapper (no need to change working directory - unwRapper does it for you)
unwRapper()Choose the location of the .feather files directory
As long as you allowed extRactoR do what it wanted, you will only need to confirm your location
unwRapper’s message
Running unwRapper results in the creation of two new
folders
Once finished, the function suggests to change your working directory. It is recommended to allow.
If you do not allow - note that you will have to in order to
work with moleculaR()
Run moleculaR
Assuming everything went fine, and your working directory is set to moleculaR_csv_files
# Run moleuclaR
moleculaR()
Users are then presented with the option to quit, and to
generate a 3D visualization of one of the molecules in
the set. It is crucial you have such a visualization, whether you choose
to use a different software or moleculaR’s
plot_molecule

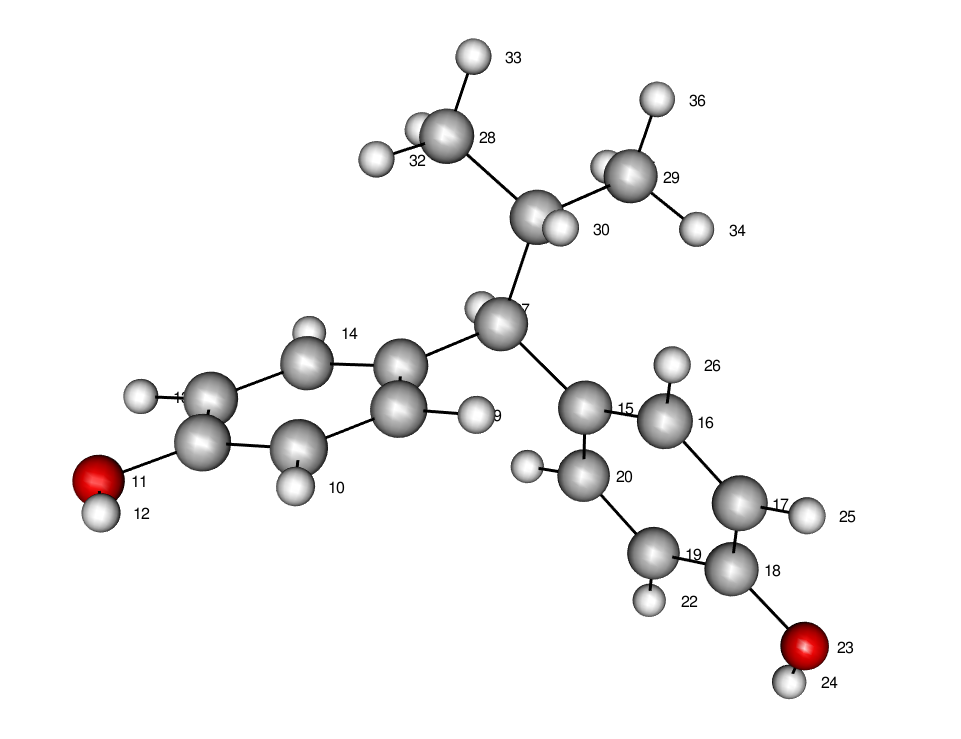
For instance, in the presented example, we have chosen to visualize Et.xyz from the Optimized_structures_xyz folder. This image will serve us in answering moleculaR’s questions - as the entire process depends on atom indexing.
Features
For each possible feature, you will be asked if you would like to use it. In cases where you choose not to, the program will move on to the next feature. The guide assumes answers to be yes, and excludes the quetion windows.
Each of the features and their options are depicted in detail in
Features - Definitions and Practice.
steRimol - B5, B1, L, loc.B1 and loc.B5 (the location of B1 and B5 vectors along the primary axis L)
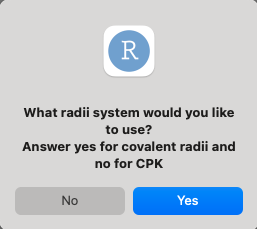
There are two radii systems implemented in this version, the first being Pyykko’s covalent radii and the second being CPK (VDW radii). The default is set to covalent radii as it holds a definituve value for all elements of the periodic table, while CPK is only defined for a small subset of them.
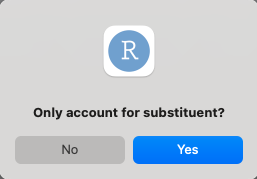
Should steRimol account for parts of the molecule that are not an extension of the primary axis? Namely, steRimol uses a graph-based “cut” of atoms that are not directly bonded to the defining axis. This is set to default as it is the generl case.
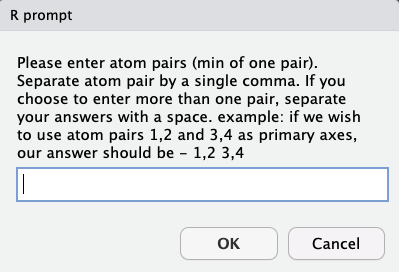
steRimol allows for the use of either a singal primary axis or several primary axes simultaneously:
User should input primary axis atoms separated by a comma, and different primary axes separated by a space.
For instance, in this example, we can imagine a scenario in which bonds 1-2, 1-3 and 1-15 can be of interest, so our answer should be 1,2 1,3 1,15
NBO (or NPA) charges - atomic values and differences
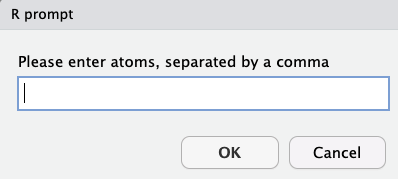
Simple usage, with hardly any explanation needed. answers should be atoms separated by a comma.
In cases where user chooses to use NBO charges, they will be preseted with the option to also use the difference in charge between atoms.
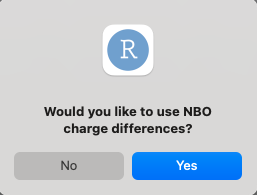
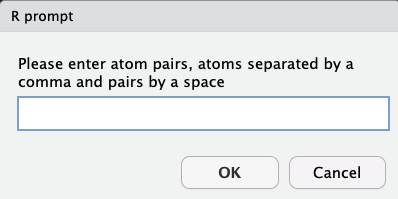
Answers should be pairs of atoms, with each pair of atoms separated by a comma, and a space between different pairs.
Dipole moments - Directly extracted from Gaussian or manipulated
Possible manipulations include:
Change of coordinate system - dipole’s vector components with respect to a coordinate system of choice allows the use of the dipole’s components as independent features
Given that the coordinate system was changed - should the origin be the center of mass of a subset of atoms (user defined)? This classifies as an esoteric use, and is recommended to use with attention to definitions.
Given that the coordinate system was changed, and that option 2 wasn’t used - should the origin of the coordinate system be the center of mass of the “basic” structure? This classifies as an esoteric use, and is recommended to use with attention to definitions.
In most cases - the coordinate system will be changed, but options 2 and 3 will be left out.
Assuming users wish to use the dipole moment as a feature, they will be presented with the option to manipulate it:
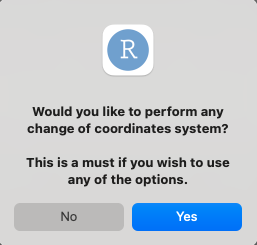
Answering “no” will leave the original dipole moment, as it given in Gaussian, which means taken with respect to the molecule’s center of mass (serving as the origin).
It is stated that this is a must in case you wish to use any of the options, as they require the transformation of coordinate systems. the y axis and the xy plane will remain as user defines them, while the only thing changing is the origin.
answering “yes” will generate the prompt
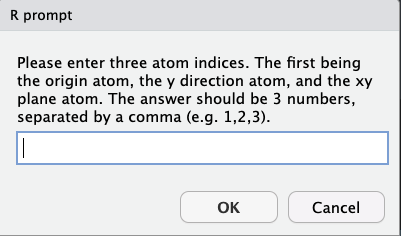
answers are given in one of two forms:
-
3 atoms, separated by a comma - define:
- atom 1 - the origin
- atom 2 - the y direction
- atom 3 - the xy plane
-
4 atoms, separated by a comma - define:
- atoms 1 and 2 - center point between the two is the origin
- atom 3 - the y direction
- atom 4 - the xy plane
Following the definition of coordinate system, user is prompted with:
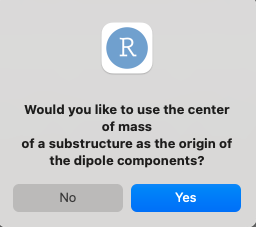
Which, in case answered “yes” will request the user to define:
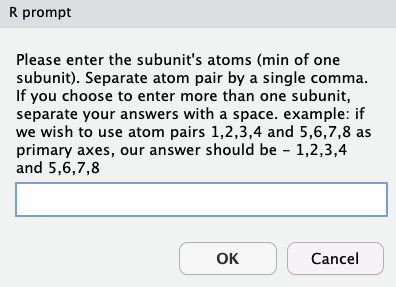
This option allows the user to use several substructures as origins, while reatining the y diretion and xy plane definitions. To use one or more substructures, sets of atoms of each substructure should be separated by a comma, and different subsets should be separated by a space.
Otherwise, if user chooses to pass this option, another option will be presented:
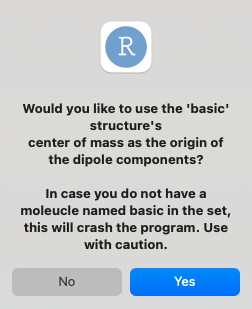
NOTE - this requires the presence of a file named basic.log, which can be whatever you find to be the “baseline” structure. it is usually the case that the smallest molecule that still holds the common substructure will serve as that structure
Vibrational Frequencies - Stretch, bend and ring vibrations
moleculaR includes the identification and extraction of vibrational frequencies that reoccur throughout sets of molecules.
These include: 1. Bonded atoms stretching vibrations 2. Two characteristic ring vibrations (only for 6 member rings) 3. Bending vibrations of two atoms that share a center atom (mostly hydrogens)
Bond Stretch
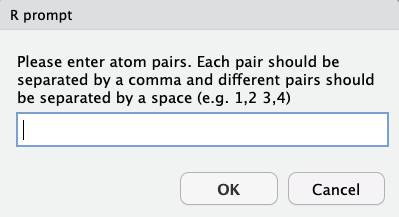
User should enter pairs of atoms, with pair atoms separated by a comma and different pairs separated by a space. Note that atoms must be bonded, and that submitting non-bonded atoms will crash the program with an error message.
Ring Vibrations
Ring vibrations are defined with the ring’s six atoms, in an ordered fashion.
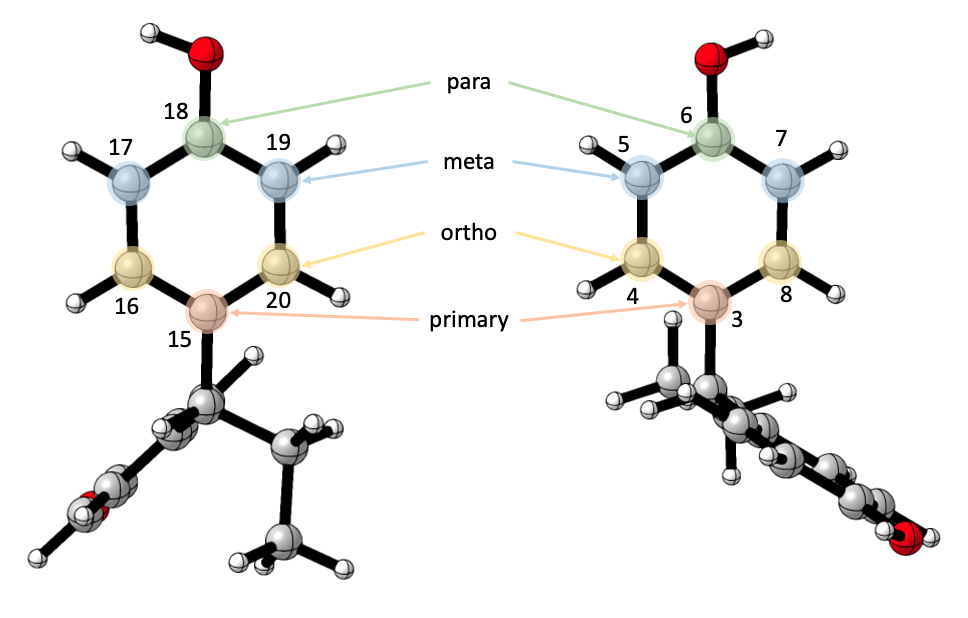
Users arbitrarily choose a “primary” atom - it is most convenient to choose the first atom on the ring, that connects the ring to the common substructure, though it really doesn’t matter. Once this atom is defined, the rest are relative to it, the one directly opposite to it is the “para” atom, the two next to is are the “ortho” atoms, and the rest are the “meta” atoms.
Note that it is possible to extract vibrations for as many rings wanted, with similar rules on input as before - same ring atoms, by the defined order, separated by a comma, and different rings separated by a space.
In this instance, our answer would have been 6,3,4,8,5,7 18,15,16,20,17,19 with 6,3,4,8,5,7 for the ring on the right, and 18,15,16,20,17,19 for the ring on the left.
Be patient and make sure you do this correctly, as series that break this rule will work, but will produce wrong results.

Angles
answers are given in one of two forms:
3 atoms, separated by a comma - define an angle created between 3 atoms
4 atoms, separated by a comma - define a dihedral between two bonds
Users may insert as many triads/quartets as they wish.


Producing Results
moleculaR will note it is done with its request to name the output file, followed by choosing the location in which you want to save both the output and the resulting inputs file - which can be used in future extractions.
Using moleculaR.input()
# To use an input file resulting from moleculaR(), do one of the following:
# 1. Explicitly indicate the path to the input file as an argument
moleculaR.input('path/to/input/file.RData')
# 2. Run the function with no arguments and choose the file
moleculaR.input()